Q-omics provides the consensus-scored SUPT16H profile across patient tissues and cancer cell-line models. SUPT16H expression is associated with patient survival in 23 of 34 cancer types, with the highest sampling consensus in ACC. Among the 18 cancer types available for tumor–normal comparison, SUPT16H is differentially expressed in 15, with the highest sampling consensus in HNSC. Additionally, SUPT16H protein abundance shows 30,280 significant protein co-abundance associations, with the highest sampling consensus in LSCC. Together, these results highlight ACC, HNSC, and LSCC as cancer lineages where SUPT16H shows reproducible signals across survival, tumor–normal expression, and patient cross-omics analyses.
Every result is evaluated using two consensus scores. Sampling consensus measures how consistently a finding is reproduced within a cancer lineage across different conditions. Lineage consensus measures how broadly the result is shared across cancer types, distinguishing pan-cancer signals from lineage-specific patterns.
Premium analyses for SUPT16H — synthetic lethality, tumor antigen, and pembrolizumab response.
This table summarizes SUPT16H survival associations across molecular data types. SUPT16H RNA expression shows survival associations in the most cancer types (23), followed by mutation status (9) and mass-spec protein abundance (6). The rightmost column indicates the cancer type with the highest sampling consensus for each molecular layer.
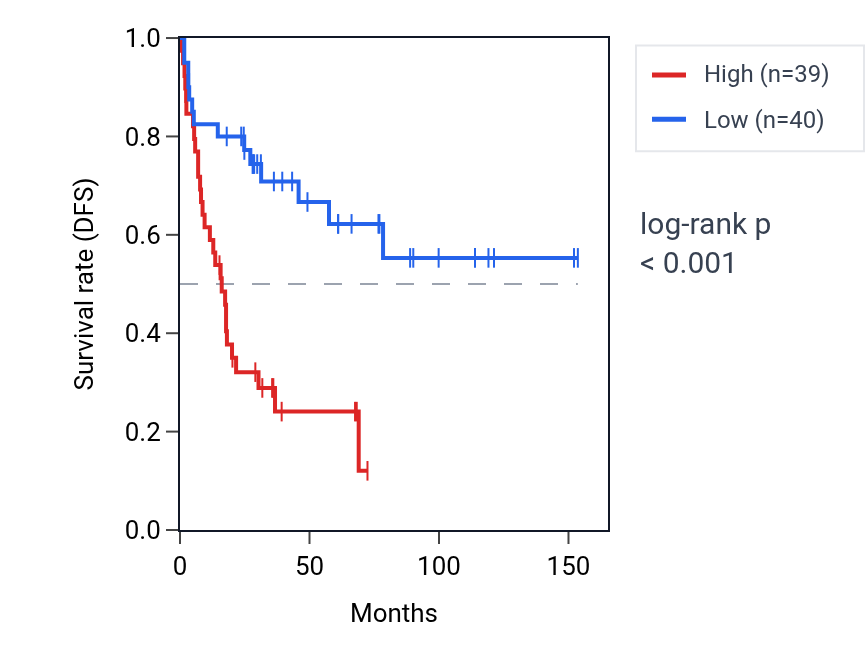
This table ranks reproducible SUPT16H RNA expression–survival associations across cancer types. High SUPT16H expression shows unfavorable associations in ACC, LIHC, BLCA and KICH, but favorable associations in KIRC and BRCA. The ACC Kaplan–Meier curve shows clear separation, with the high-expression group declining faster, consistent with the unfavorable association (log-rank p < 0.001). Together, the overview and detailed table identify ACC as the clearest survival context for SUPT16H RNA expression.
This table summarizes SUPT16H tumor–normal expression differences by data type. RNA shows broader differences across cancer types, with a lineage consensus of 15, while mass-spec protein shows differences in 7. The strongest signals are observed in HNSC for RNA and COAD for protein.
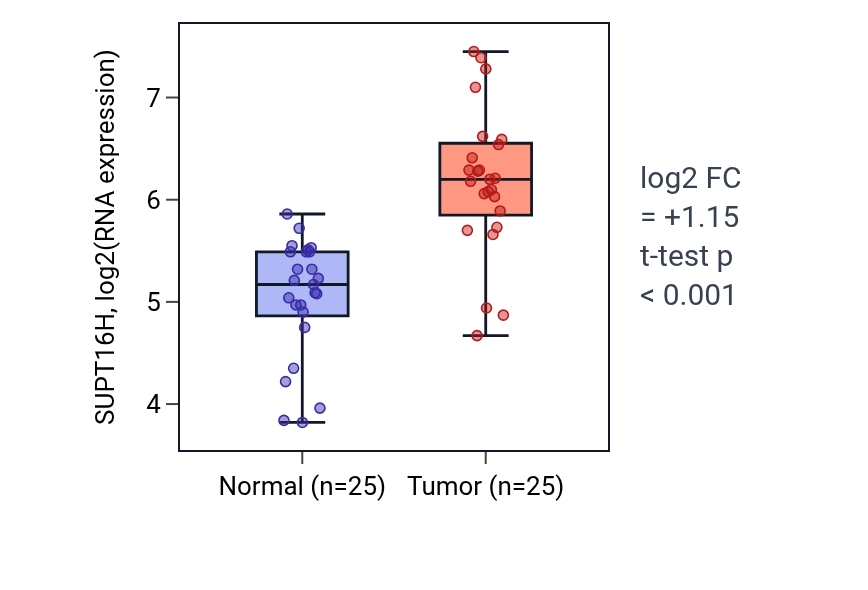
This table ranks reproducible tumor–normal expression differences for SUPT16H. A negative fold-change indicates higher expression in normal tissue than in tumor tissue. SUPT16H shows higher tumor expression in HNSC, STAD, LIHC, COAD, LUSC and BLCA. The HNSC box plot shows higher SUPT16H RNA expression in tumor versus normal tissue (log2 FC = +1.147, t-test p < 0.001).
This table shows molecular features associated with SUPT16H in patient tissues and cancer cell lines. In patient samples, SUPT16H shows the broadest associations at the RNA and protein expression levels, with LSCC recurring as the lineage with the largest associated feature set. In cancer cell lines, SUPT16H RNA and mutation anchors are most strongly linked to RNA-expression features, especially in OVARY, while CRISPR and shRNA rows add functional-dependency signals in CNS and BLOOD_Leukemia.